
More Information
Submitted: December 10, 2023 | Approved: December 26, 2023 | Published: December 27, 2023
How to cite this article: Razali RR, Lubis MA. Apert Syndrome: A Case Report. Clin J Obstet Gynecol. 2023; 6: 233-235.
DOI: 10.29328/journal.cjog.1001154
Copyright License: © 2023 Razali RR, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Apert syndrome; Craniosynostosis; Syndactyly

Apert Syndrome: A Case Report
Renardy Reza Razali1 and Munawar Adhar Lubis2*
1Departement of Obstetrics and Gynecology, Faculty of Medicine, Riau University, Pekanbaru, Indonesia
2Resident of Obstetrics and Gynecology, Faculty of Medicine, Riau University, Pekanbaru, Indonesia
*Address for Correspondence: Munawar Adhar Lubis, Resident of Obstetrics and Gynecology, Faculty of Medicine, Riau University, Pekanbaru, Indonesia, Email: [email protected]
Background: Apert syndrome is a type 1 acrocephalosyndactyly, a rare syndrome characterized by the presence of multiple craniosynostoses, dysmorphic facial manifestations, and syndactyly of hand and feet. It affects 1:100.00 of birth and is the second most common of syndromic craniosynostosis. Molecular genetic tests that identify the heterozygous pathogenic variant in FGFR2 genes - identical with Apert syndrome cost too high to be applicable in developing countries. Therefore, the diagnosis of Apert syndrome should be suspected from the clinical findings. Three cases from the Community of Indonesian Apert Warrior Group were collected. These series were based on medical and surgical records. We obtained the patient characteristics from the phenotypic manifestations only.
Case report: We present a case of a newborn baby girl, with similar anatomical findings, such as skull shape abnormality, midface hypoplasia, intraoral disfigurement, and hands and feet deformities that resemble Apert Syndrome. Our series presents similar Apert syndrome characteristics, such as typical craniofacial dysmorphic with symmetrical syndactyly of both upper and lower extremities. These clinical findings are essential to establish an initial diagnosis of Apert Syndrome.
Apert syndrome was first discovered by Wheaton in 1894. In 1906, Dr. Eugene Charles Apert published a summary of nine cases. Aperts syndrome, or acrocephalosyndactylia, is a developmental malformation characterised by cranio-synostosis, a cone-shaped calvarium, midface hypoplasia, pharyngeal attenuation, ocular manifestations, and syndactyly of the hands and feet. According to Cohen, 15 out of 10 lakh live births suffer from Apert syndrome. Apert syndrome is caused by a mutation in the FGFR2 gene on chromosome 10 [1-3]. The FGFR2 gene produces a protein called fibroblast growth factor receptor; one of its functions is to signal immature cells to become bone cells in the developing embryo or fetus. Mutations in specific parts of FGFR2 will change this protein and cause prolonged signalling, resulting in premature fusion of the bones of the skull, fingers, and toes. 2, 3, and 4 FGFR2 gene mutations can be inherited from parents who suffer from Apert syndrome with an autosomal dominant pattern, or it could also be a new spontaneous mutation [1,4]. Previous studies have shown an association between older paternal ages and Apert syndrome. Apert syndrome occurs around 1 in 160,000– 200,000 live births and affects men and women equally. The main characteristics of Apert syndrome are malformation of the skull, retrusion of the midface, fusion of the fingers and toes, brahicephaly, acrocephaly, flat back of the head, prominent eyes, and strabismus. Manifestations of Apert syndrome in the cavity mouth in the form of a prominent mandible, drooping corners of the mouth, cleft palate, tooth malposition, crowding, delayed tooth eruption, thick alveolar ridge, malocclusion, and arch deformity of the hard palate called Byzantine arch deformity [3]. 3.5 In a normal child, the skull bones are formed from several “plates” that remain separated from each other and then gradually grow together to form the adult skull. In children with Apert syndrome, premature fusion of these plates occurs (craniosynostosis), which causes retardation of brain growth and increased pressure in the brain as the brain grows [2,3]. The diagnosis of Apert syndrome can be made from a clinical examination and X-ray photographs of the skull, which will show premature closure of the skull. The combination of craniofacial problems and fused fingers and toes is what differentiates Apert syndrome from other similar disorders. A genetic examination can also be carried out to further confirm the diagnosis [3]. Management of children with Apert syndrome requires a team approach consisting of a craniofacial surgeon, neurosurgeon, oral surgeon, ENT specialist, audiologist, speech pathologist, psychologist, ophthalmologist, pedodontist, and orthodontist [4]. The main method of treatment is surgery. This surgery is needed to correct craniofacial abnormalities and fused fingers and toes. Apart from surgery, treatment is also aimed at correcting the upper respiratory tract, eye abnormalities, or abnormalities affecting the teeth [5].
A 39-year-old multiparous woman with a gestation period of 38 weeks came to the emergency room at Arifin Achmad Pekanbaru Regional Hospital with complaints of wanting to have a hard time. An examination showed maximal cervical dilatation, the foetal head was palpable down at Hodge 4, and the FHR was 150 bpm regular. A baby girl was born in the emergency room at Arifin Achmad Hospital Pekanbaru. She is the second child from a nonconsanguineous marriage and has one normal sibling, and the mother gave birth normally without a history of trauma, infection, or drug use during that time. No family history of similar complaints or other congenital abnormalities was reported. Examination reveals an abnormal turribrachycephalic head contour (high and shortened AP), flat occiput, and prominent frontal region. There is ocular proptosis, strabismus, hypertelorism, and a lateral palpebral fissure that shifts downward. She has a depressed nose bridge and a thick nose with a rounded tip and crossbow-shaped lips. The patient had a midfacial deficiency with a hypoplastic and retruded maxilla (Figure 2). Bilaterally symmetrical syndactyly with complete fusion of the five fingers of both hands with the thumbs placed inward (Figure 3), there is also both feet with deformation of the big toes. Fused fingers and toes have separate nails. Intraoral, no teeth, V-shaped maxillary arch, and pseudocleft palate (Figure 1). There were no other obvious congenital malformations, and a systemic examination showed no other abnormalities. We carried out a chromosome examination and obtained suspicious results for FGFR2 gene mutations, which were consistent with Apert syndrome. The baby has been consulted by a paediatric surgeon, but palate reconstruction is only planned at the age of 3 years (Figures 1-5).
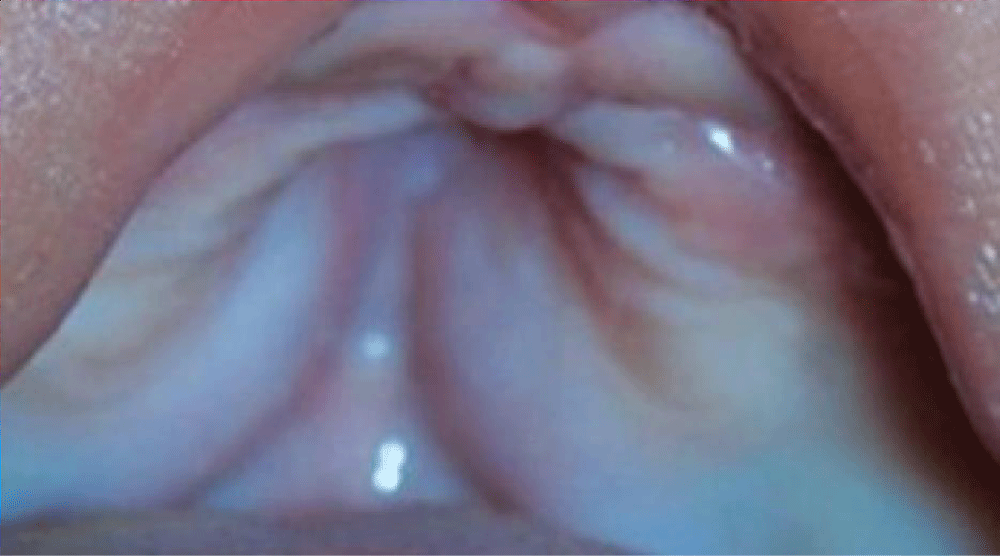
Figure 1: Palatum Deformity.
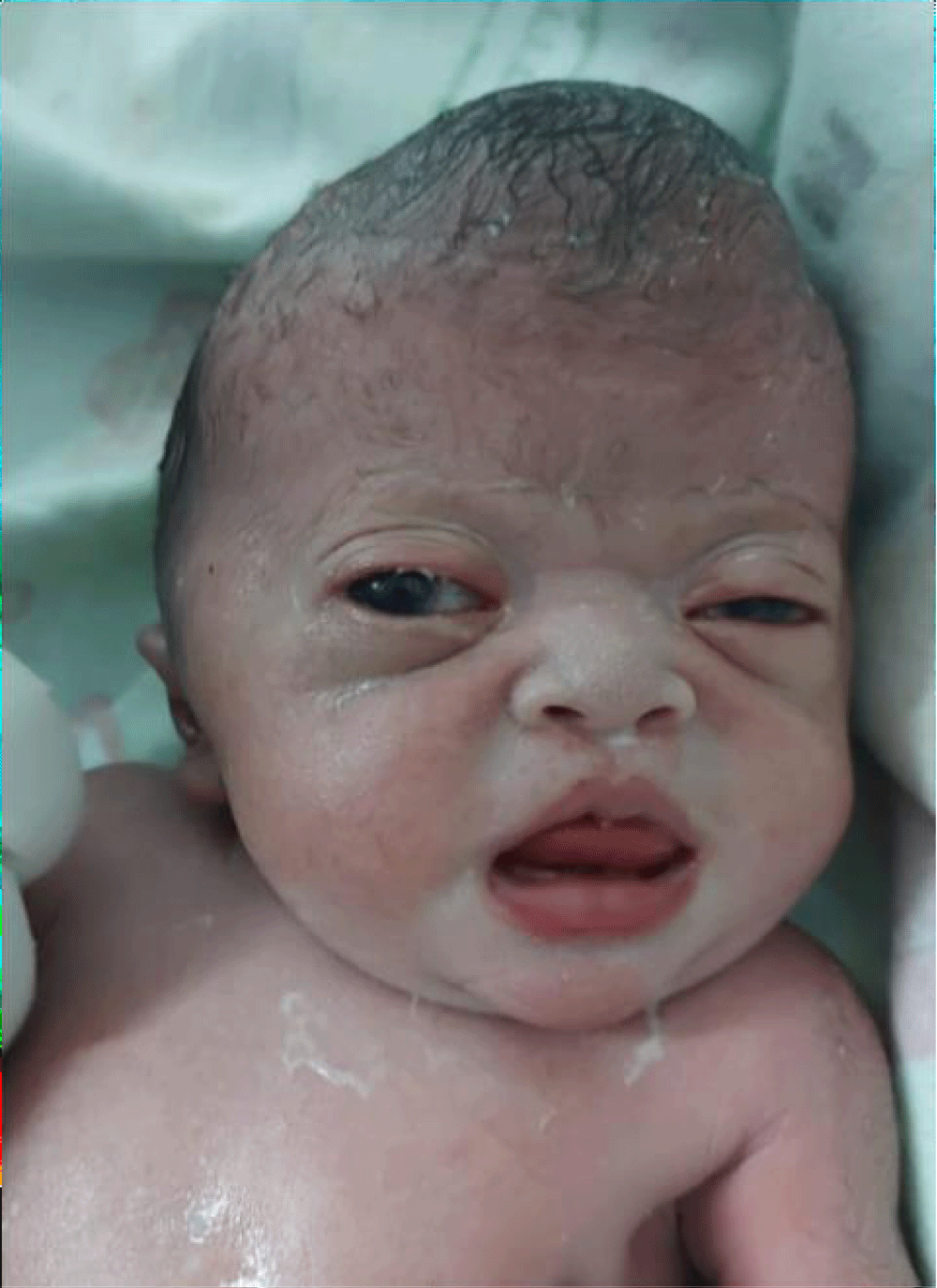
Figure 2: Baby facial profile.
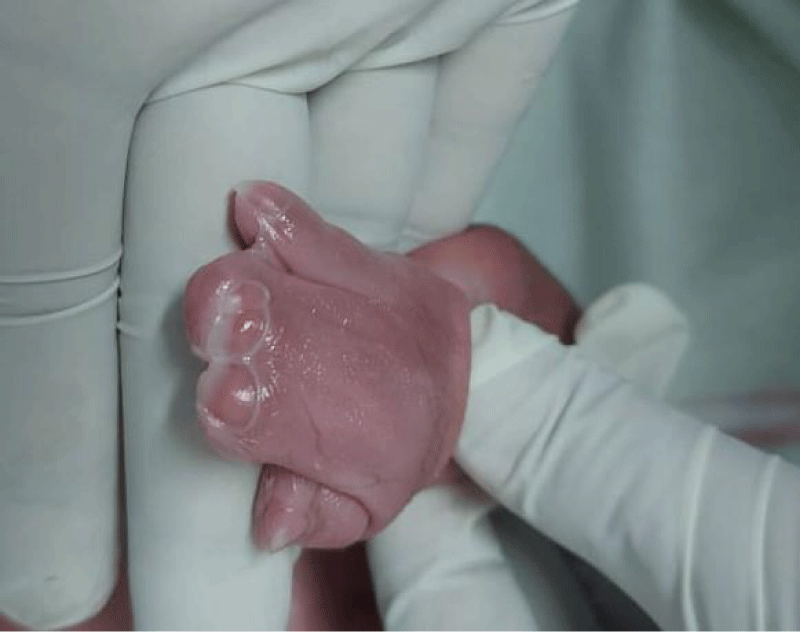
Figure 3: Connected Fingers and Wide Thumb.
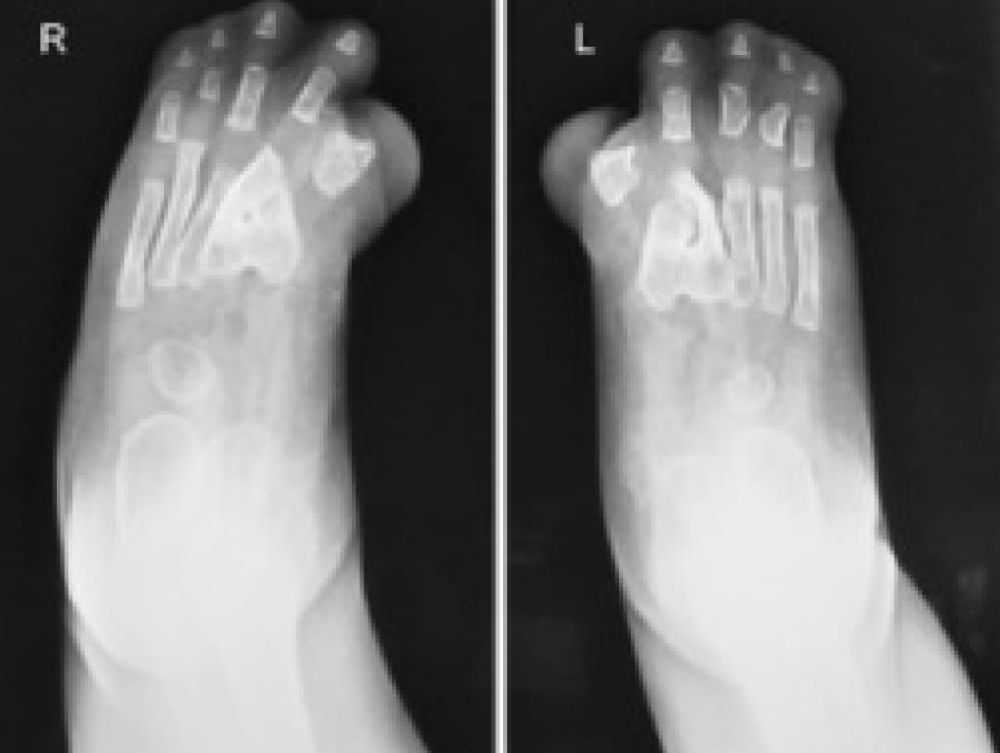
Figure 4: Foot X-ray Results.
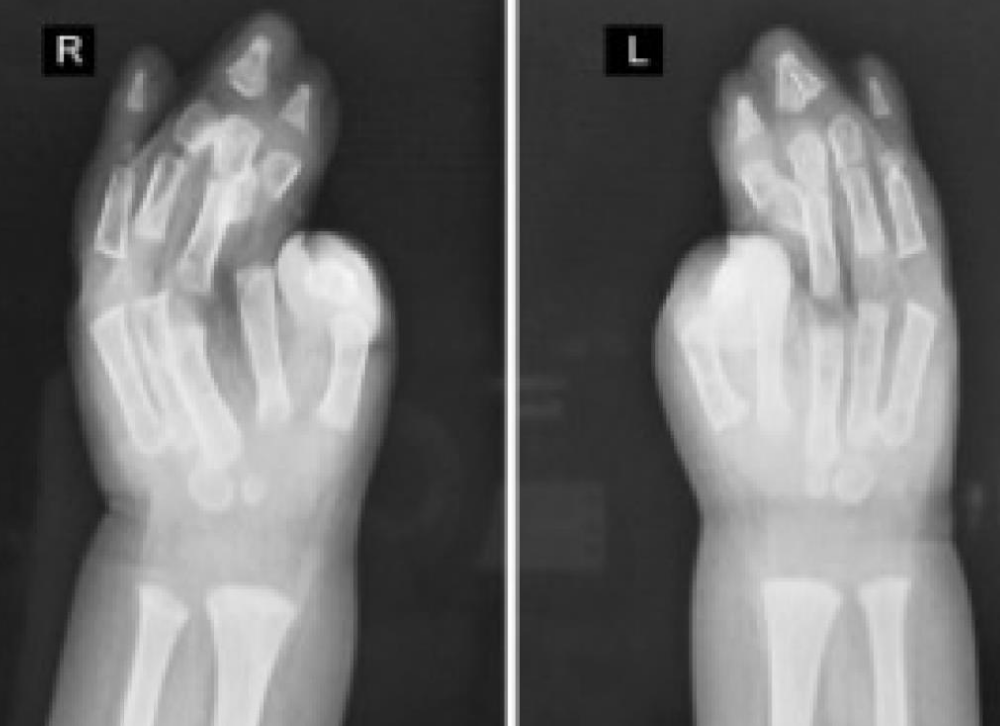
Figure 5: Hand X-ray Results.
Apert syndrome is an autosomal dominant disorder caused by a mutation in fibroblast growth factor receptor-2 (FGFR-2) on chromosome 10q. Suture progenitor cells with mutated FGFR-2 are unable to transduce signals from extracellular FGF, and as a result, they do not produce the fibrous material necessary for a normal calvarial suture. Most cases are sporadic, due to new mutations influenced by the father's age [6,7]. Prodromal features of the typical turribrachycephalic head shape are early craniosynostosis of the coronal sutures and agenesis of the saggital and metopic sutures, resulting in a wide defect extending from the glabella to the posterior fontanel. Also, spheno-occipital and sphenoethmoidal synchondrosis and early fusion of the frontoethmoidal suture cause the anterior and posterior skull bases to shorten, resulting in a reduction in the height of the pharynx [8,9]. Premature fusion of the sutures with ongoing brain growth can lead to increased intracranial pressure, which can be seen as increased convolutional signs on skull radiographs. This in turn results in a hypoplastic midface and a vertically prominent craniofacial complex. Ocular proptosis, downward tilt of the lateral canthus and palpebral fissure (antimongoloid oblique), and hypertelorism occur due to the shortening of the bony orbit. There is a depressed nasal bridge with a deviated nasal septum. Hypoplastic and retropositioned maxilla. The lips are arc-shaped and often cannot form a lip seal. Syndactylia, or finger adhesions, cause immobility of the fingers due to ossification of the interphalangeal joints due to segmentation of the embryonic phalanges. The involvement of the first or fifth toe in this bone mass varies [10,11]. There may be similar deformities involving the feet (glove hands and sock feet). High intraoral arch and sagittally narrow palate are seen with lateral palatal swelling with a prominent central fissure and are mostly present with pseudopalate. The maxillary arch is V-shaped and narrows sagittally. Severe crowding of teeth, delayed tooth eruptions, and thick gingiva are common features. The posterior tilt of the maxilla can cause class III malocclusion in the future [12]. Central nervous system abnormalities (megalocephaly, pyramidal tract abnormalities), skeletal abnormalities (limited mobility of the glenohumeral joint, elbow joint, etc.), cardiovascular, genitourinary and gastrointestinal, mental retardation, visual and auditory, and speech defects have also been noted. The literature also reports skin manifestations in Apert syndrome, such as acne, hyperhidrosis, hypopigmentation, and plantar surface hyperkeratosis [11]. According to the literature, Apert and Crouzon syndrome appear to be the same syndrome, with the exception of syndactyly of the hands and feet in Apert syndrome. A cleft or pseudocleft palate is frequently found in Apert syndrome, whereas these features are very rare in Crouzon syndrome [12,13]. Treatment of Apert syndrome begins at birth, and a multidisciplinary approach is required to arrive at a collaborative corrective plan for the deficiency. Craniectomy is often performed at 6 months of age to treat craniosynostosis. Corrective surgery for syndactyly is performed in the first year of life and completed by 3 to 4 years of age [14]. Cosmetic correction of midface and pseudocleft deficiencies occurs at 4 to 6 years of age. Orthodontic and orthognathic surgery is performed after permanent tooth eruption and growth are complete. Nonsurgical manipulation of Apert syndrome may be a possibility in the future, for example, by using selective inhibitors of the FGFR-kinase domain. Genetic counselling is an important factor because the risk of recurrence for an affected individual to have affected offspring is 50% [15,16].
Apert syndrome is a rare disorder that can be detected during pregnancy by ultrasound examination. an autosomal dominant disorder caused by a mutation in fibroblast growth factor receptor-2 (FGFR-2) on chromosome 10q. A chromosome examination is needed to confirm the diagnosis of Apert syndrome.
- Cohen MM Jr. Apert syndrome. Orphanet J Rare Dis. 2017 Nov 14;2:6. doi:10.1186/1750-1172-2-6.
- Wilkie AO, Slaney SF, Oldridge M, Poole MD, Ashworth GJ, Hockley AD, Hayward RD, David DJ, Pulleyn LJ, Rutland P, et al. Apert syndrome results from localized mutations of FGFR2 and is allelic with Crouzon syndrome. Nat Genet. 1995 Feb;9(2):165-72. doi: 10.1038/ng0295-165. PMID: 7719344.
- Lajeunie E. Apert syndrome: a review of clinical features and mutations in the FGFR2 gene. Int J Oral Maxillofac Surg. 2019 Apr;28(2):95-102.
- Anderson PJ. Cognitive and educational outcome of craniosynostosis. Plast Reconstr Surg. 2014 May;125(5):e463-70.
- Sowińska A. Apert syndrome: analysis of clinical features, genetics, and outcomes of 28 Polish patients. J Appl Genet. 2017 May;59(2):179-85.
- Lattanzi W. Long-term follow-up of a patient affected by Apert syndrome: a case report. J Med Case Rep. 2018 May 5;12(1):116.
- Sharma VP. Apert syndrome: a report of two cases. J Clin Diagn Res. 2014 Jul;8(7):ZD29-31.
- Baroni T. Apert syndrome: report of two cases in the same family and literature review. Arq Neuropsiquiatr. 2012 Sep;70(9):730-2.
- Holmes G. Functional outcomes in Apert syndrome. Plast Reconstr Surg. 2012 Feb;129(2):187e-93e.
- Yamaguchi KT. Apert syndrome: report of two cases. Einstein (Sao Paulo). 2014 Jan-Mar;12(1):106-10.
- Moloney DM, Slaney SF, Oldridge M, Wall SA, Sahlin P, Stenman G, Wilkie AO. Exclusive paternal origin of new mutations in Apert syndrome. Nat Genet. 1996 May;13(1):48-53. doi: 10.1038/ng0596-48. PMID: 8673103.
- Cohen MM Jr, Kreiborg S. Skeletal abnormalities in the Apert syndrome. Am J Med Genet. 1993 Oct 1;47(5):624-32. doi: 10.1002/ajmg.1320470509. PMID: 8266987.
- Anderson PJ, Hall CM, Evans RD, Hayward RD, Jones BM. The facial morphology in Apert syndrome: a study of cephalometric and anthropometric measures. J Craniofac Surg. 2015 Jan;8(1):9-12.
- Slaney SF, Oldridge M, Hurst JA, Moriss-Kay GM, Hall CM, Poole MD, Wilkie AO. Differential effects of FGFR2 mutations on syndactyly and cleft palate in Apert syndrome. Am J Hum Genet. 1996 May;58(5):923-32. PMID: 8651276; PMCID: PMC1914616.
- Kimonis V, Gold JA, Hoffman TL, Panchal J, Boyadjiev SA. Genetics of craniosynostosis. Semin Pediatr Neurol. 2007 Sep;14(3):150-61. doi: 10.1016/j.spen.2007.08.008. PMID: 17980312.
- Yu JC, Lajeunie E, Tyndale-Biscoe A, Hayward R, Jones BM, Oldridge M, Salonen R, Walsh S, Slaney SF, Quarrell OW, Wilkie AO. Apert syndrome: correlation between phenotype and genotype. Am J Med Genet. 2017 Oct 23;57(4):523-30.